Snakemake example
You can write and run unit tests for bioinformatics pipelines just like you would for regular software. While testing is strongly encouraged for re-usable pipelines in particular, sometimes unit testing is not a high priority, such as for one-off analysis projects. A more basic check you can do is make sure your pipeline has correct syntax. For Snakemake, you can dry-run and lint the Snakemake workflow to ensure it passes minimal quality standards.
Take a look at the example Snakemake workflow:
snakemake-workflow/workflow/Snakefile
rule echo:
output:
"output.txt"
container: "nciccbr/minimal:v1"
shell:
"""
echo "Hello, world!" > {output}
"""We’ll use a GitHub Actions workflow to check that our Snakemake workflow can complete a dry-run and that it passes Snakemake’s linting checks.
.github/workflows/build-python.yml
name: snakemake
on:
push:
branches:
- main
- develop
pull_request:
jobs:
dryrun-lint:
runs-on: ubuntu-latest
1 container:
image: snakemake/snakemake:v7.32.4
steps:
- uses: actions/checkout@v4
2 - name: Dry-run
run: |
snakemake \
-s ./snakemake-workflow/workflow/Snakefile \
--dry-run
3 - name: Lint
run: |
snakemake \
-s ./snakemake-workflow/workflow/Snakefile \
--lint- 1
- Specify a Docker container for all steps in this job to run inside.1
- 2
- Dry-run the Snakemake workflow. If the dry run completes successfully, you at least know there aren’t syntax errors in your Snakemake code.
- 3
- Lint the workflow with Snakemake’s built-in linter to ensure code quality.
GitHub’s standard runners are genearlly limited to 4 CPUs and 16 GB RAM2. You won’t be able to run a fully-featured bioinformatics pipeline on real data in GHA.
Add this new GHA workflow to your repo and see what happens. Do both the dry-run and lint steps complete successfully? Do any changes need to be made to the Snakemake workflow make these steps pass?
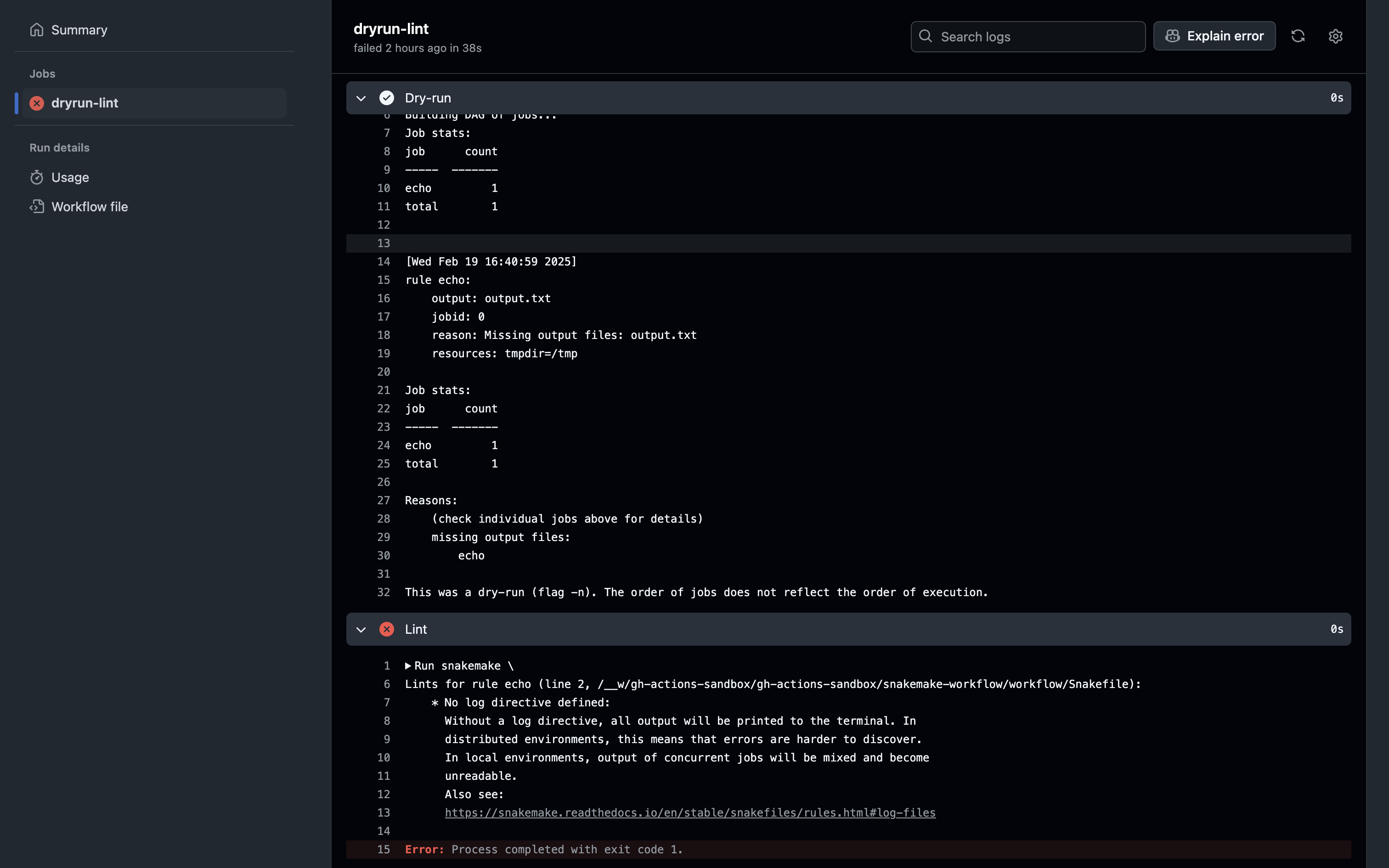
The dry run succeeded, but the lint step failed because we didn’t define a log file for the rule in the Snakefile. Let’s modify it:
.github/workflows/build-python.yml
rule echo:
output:
"output.txt"
container: "nciccbr/minimal:v1"
log: "log/echo.log"
shell:
"""
echo "Hello, world!" > {output} 2> {log}
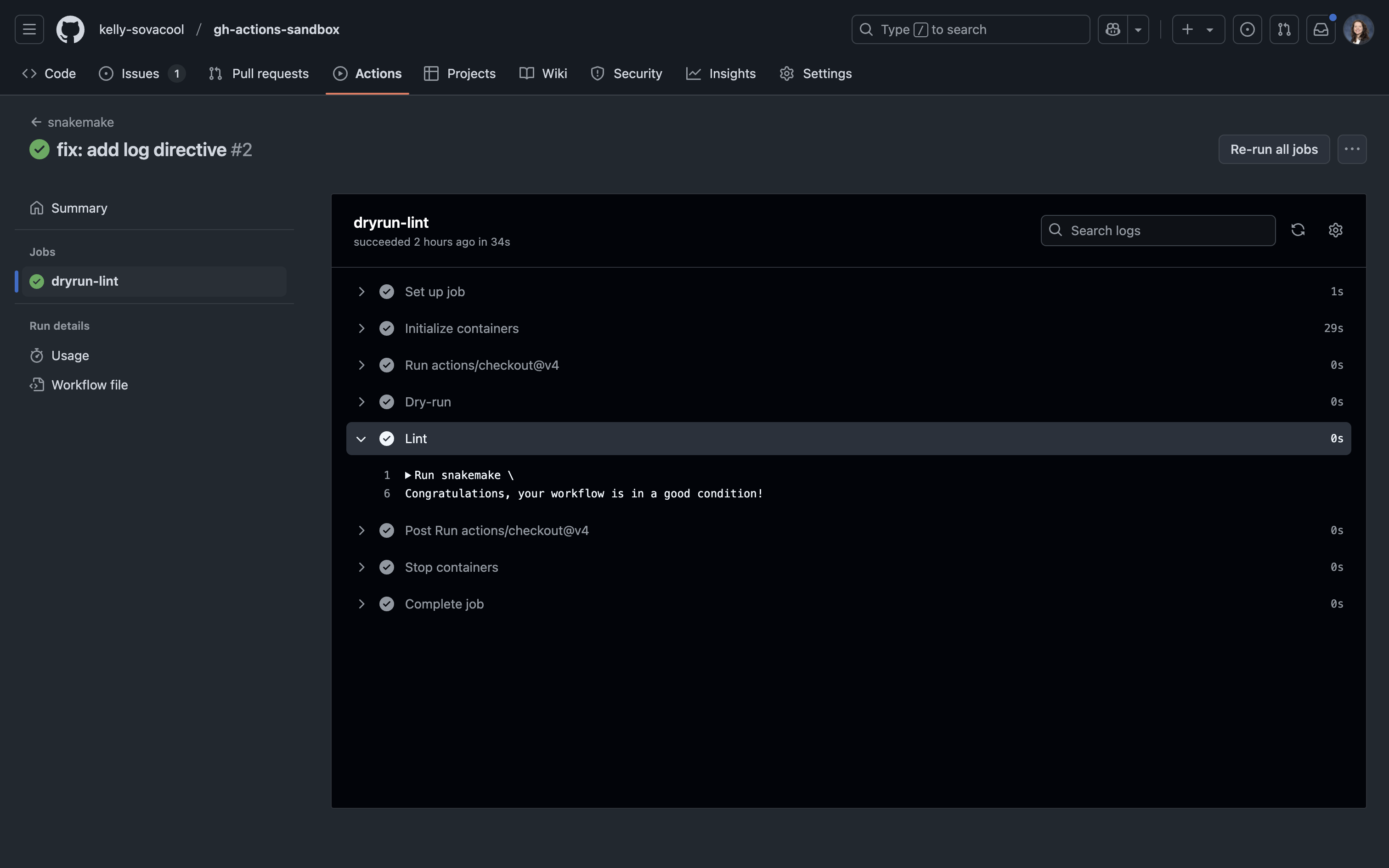
"""Add, commit, and push this change to your repo. Now the lint step passes and tells you that the Snakemake workflow is in good condition:
More workflows for bioinformatics pipelines
- Snakemake (CCBR) https://github.com/CCBR/actions/blob/main/examples/build-snakemake.yml
- Nextflow (CCBR) https://github.com/CCBR/actions/blob/main/examples/build-nextflow.yml
- Nextflow (nf-core) https://github.com/nf-core/rnaseq/blob/master/.github/workflows/ci.yml
Footnotes
Running jobs in a container: https://docs.github.com/en/actions/writing-workflows/choosing-where-your-workflow-runs/running-jobs-in-a-container↩︎
Resources for standard GitHub runners: https://docs.github.com/en/enterprise-cloud@latest/actions/using-github-hosted-runners/using-github-hosted-runners/about-github-hosted-runners#standard-github-hosted-runners-for-public-repositories↩︎